





(C) 2013 Jephris Gere. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
For reference, use of the paginated PDF or printed version of this article is recommended.
Citation: Gere J, Yessoufou K, Daru BH, Mankga LT, Maurin O, van der Bank M (2013) Incorporating trnH-psbA to the core DNA barcodes improves significantly species discrimination within southern African Combretaceae. In: Nagy ZT, Backeljau T, De Meyer M, Jordaens K (Eds) DNA barcoding: a practical tool for fundamental and applied biodiversity research. ZooKeys 365: 127–147. doi: 10.3897/zookeys.365.5728
Recent studies indicate that the discriminatory power of the core DNA barcodes (rbcLa + matK) for land plants may have been overestimated since their performance have been tested only on few closely related species. In this study we focused mainly on how the addition of complementary barcodes (nrITS and trnH-psbA) to the core barcodes will affect the performance of the core barcodes in discriminating closely related species from family to section levels. In general, we found that the core barcodes performed poorly compared to the various combinations tested. Using multiple criteria, we finally advocated for the use of the core + trnH-psbA as potential DNA barcode for the family Combretaceae at least in southern Africa. Our results also indicate that the success of DNA barcoding in discriminating closely related species may be related to evolutionary and possibly the biogeographic histories of the taxonomic group tested.
DNA barcoding, closely related species, Combretaceae, southern Africa
Combretaceae is a medium-sized family within Myrtales, comprising about 500 species in 17 to 23 genera. It has long been referred to as a complex phylogenetic and taxonomic group (
The major distinguishing feature of the family is the presence of unicellular combretaceous hairs on the abaxial leaf surfaces, a diagnostic trait in many other species of Myrtales and even beyond the group e.g. the Cistaceae Juss. family, tribe Cisteae (

Selected inflorescences of seven Combretum species indicating closely related species evaluated based upon floral characters. A Combretum paniculatum B Combretum microphyllum C Combretum platypetalum D Combretum hereroense E Combretum apiculatum F Combretum molle G Combretum kraussii. All photographs by O. Maurin.

Selectedmature dry four-winged fruits of closely related species of genus Combretum. A Combretum mkuzense B Combretum microphyllum C Combretum englerii D Combretum apiculatum E Combretum moggii F Combretum albopunctatum G Combretum collinum. All photographs by O. Maurin.
Here, we propose that DNA barcoding may provide such a complementary tool to ease species delimitation within the group. DNA barcoding involves the use of a short and standardised DNA sequence that can help assign, even biological specimens devoid of diagnostic features, to species (
In this study, we evaluated the efficacy of DNA barcoding as a tool to augment morphological species discrimination within Combretaceae. Specifically, we (1) assessed the potential of four markers to discriminate southern African species of the family, and (2) assessed the efficacy of barcodes across major clades including subgenera and sections within the largest genus Combretum.
Sampling includes one to six accessions of 58 species out of the 63 species representing the six genera of Combretaceae in southern Africa. These genera include Combretum (43 species included in this study), Lumnitzeria Wild. (one species included), Meiostemon Exell and Stace (one species included), and Quisqualis L. (one species included), Pteleopsis Engl. (two species included), and Terminalia (ninespecies included).
Collection details, taxonomy, voucher numbers, GPS coordinates, field pictures, and sequence data (only matK and rbcLa) are archived online on the BOLD system (www.boldsystems.org). Voucher information, name of herbarium, GenBank and BOLD accession numbers are listed in Appendix 1.
Genomic DNA was extracted from silica gel-dried and herbarium leaf material following a modified cetyltrimethyl ammonium bromide (CTAB) method of
All PCR reactions were carried out using Ready Master Mix (Advanced Biotechnologies, Epsom, Surrey, UK). We added 4.5% of dimethyl sulfoxide (DMSO) to the PCR reactions of nrITS to improve PCR efficiency. Amplification of rbcLa was done using the primer combination: 1F: 724R (
The following programme was used to amplify rbcLa and trnH-psbA: pre-melt at 94 °C for 60 sec, denaturation at 94 °C for 60 sec, annealing at 48 °C for 60 sec, extension at 72 °C for 60 sec (for 28 cycles), followed by a final extension at 72 °C for 7 min; for matK, the protocol consisted of pre-melt at 94 °C for 3 min, denaturation at 94 °C for 60 sec, annealing at 52 °C for 60 sec, extension at 72 °C for 2 min (for 30 cycles), final extension at 72 °C for 7 min. For nrITS and spacer psaA-ycf3 the protocol consisted of pre-melt at 94 °C for 1 min, denaturation at 94 °C for 60 sec, annealing at 48 °C for 60 sec, extension at 72 °C for 3 min (for 26 cycles), final extension at 72 °C for 7 min.
Purification of the amplified products was done using QIAquick columns (QIAgen, Germany) following the manufacturer’s manual. The purified products were then cycle-sequenced with the same primers used for amplification using BigDyeTM v3.1 Terminator Mix (Applied Biosystems, Inc, ABI, Warrington, Cheshire, UK). Cleaning of cycle-sequenced products was done using EtOH-NaCl, followed by sequencing on an ABI 3130xl genetic analyser.
Sequences were assembled, trimmed and edited using Sequencher v4.6 (Gene Codes Corp, Ann Arbor, Michigan, USA). Alignment was done using Multiple Sequence Comparison by Log-Expectation v3.8.31 (
Performance of DNA markers in species delimitation was tested at three taxonomic levels (family, subgenus, and section). At family level, we evaluated four single markers: rbcLa, matK, trnH-psbA, and nrITS. We also tested the core barcodes, i.e. rbcLa + matK (
Barcode gap was evaluated in two ways: (1) we compared genetic variation within species (intraspecific genetic distance) versus between species (interspecific genetic distance). This comparison was based on the mean, median, and range of both distances; (2) in addition, we also used
The discriminatory power of DNA regions was conducted using three distance-based methods including Near Neighbour, Best Close Match (
We also used the PCR success rate to evaluate the DNA regions. This evaluation was conducted based on the percentage of successful amplification.
The test for species monophyly was conducted on a Neighbour-Joining (NJ) tree. We considered that a species is monophyletic when all individuals of the same species cluster on the NJ phylogram that we reconstructed. As such, the best barcode should provide the highest proportion of monophyletic species. We then evaluated for each DNA region and concatenated regions, the proportion of monophyletic (i.e. correct identification) and non-monophyletic species (incorrect identification). All our analyses were conducted in the R package Spider v1.1-1 (
Finally, we evaluated the barcoding potential in discriminating phylogenetically deliminated clades in the phylogeny of the genus that was reconstructed based on the combination of five DNA regions (rbcL, matK, trnH-psbA, psaA-ycf3 and nrITS). The phylogeny was reconstructed based on maximum parsimony (MP) implemented in PAUP* v4.0b10 (
At subgeneric and sectional levels, we only tested the performance of core barcodes and best gene combination identified using the three criteria mentioned above (barcode gap, discriminatory power and species monophyly).
The overall characteristics of single and combined DNA regions are presented in Table 1. In general, our results indicate that the ranges and mean intraspecific distances were both lower than those of interspecific distances. Among single regions, rbcLa showed the lowest interspecific distance (mean = 0.009) with nrITS exhibiting the highest genetic variation between species (mean = 0.110). For all marker combinations, the mean interspecific distances varied between 0.011 and 0.014. Assessing the index of sequence divergence K for each region, we found that nrITS showed the highest divergence (K = 21) whereas trnH-psbA exhibited the lowest divergence (K = 3). For the combined regions, K varied between 10 and 13, with an average of 10 substitutions between sequence-pairs (Table 1).
Statistics of all gene regions for the southern African Combretaceae included in the study.
| DNA regions | No. of seq | Seq length | K | Range inter | Mean inter (±SD) | Range intra | Mean intra (±SD) | Threshold (%) |
|---|---|---|---|---|---|---|---|---|
| rbcLa | 152 | 552 | 4 | 0-0.09 | 0.009±0.012 | 0-0.08 | 0.002±0.009 | 0.04 |
| matK | 133 | 771 | 6 | 0-0.07 | 0.014±0.011 | 0-0.02 | 0.002±0.004 | 1.10 |
| trnH-psbA | 116 | 1034 | 3 | 0-0.15 | 0.047±0.035 | 0-0.03 | 0.003±0.007 | 1.80 |
| nrITS | 91 | 821 | 21 | 0-0.21 | 0.110±0.045 | 0-0.05 | 0.004±0.010 | 1.70 |
| rbcLa+matK | 129 | 1323 | 10 | 0-0.78 | 0.012±0.009 | 0-0.05 | 0.002±0.006 | 1.31 |
| rbcLa+matK+trnH-psbA | 87 | 2358 | 11 | 0-0.04 | 0.012±0.007 | 0-0.02 | 0.002±0.004 | 0.5 |
| rbcLa+matK+nrITS | 74 | 2144 | 9 | 0-0.04 | 0.011±0.006 | 0-0.02 | 0.002±0.004 | 0.70 |
| rbcLa+matK+nrITS+trnH-psbA | 70 | 3178 | 13 | 0-0.04 | 0.014±0.007 | 0-0.02 | 0.002±0.004 | 1.17 |
The distribution ranges of inter- versus intraspecific distances for all regions, showed a clear overlap between both distances (Figures 3a, b and 4), indicating the existence of a barcode gap. Comparing the smaller inter- versus the largest intraspecific distances for each region, our results further support the existence of barcode gap in all regions, but the proportion of sequences with barcode gap varied significantly with the regions tested (Table 2). Notably, the combination of all four regions exhibited the highest proportion of sequences with barcode gap (84%) followed by nrITS (73%), then core + nrITS (64%), and core + trnH-psbA (57%), with the lowest proportion found in rbcLa (13%) (Table 2).
Optimised genetic distances used as threshold for species delimitation in Best Close Match method are shown in Table 1. Apart from rbcLa (threshold = 0.04%), core + trnH-psbA (threshold = 0.5%) and core + nrITS (threshold = 0.7%), the thresholds for the remaining single and gene combinations were greater than 1%.
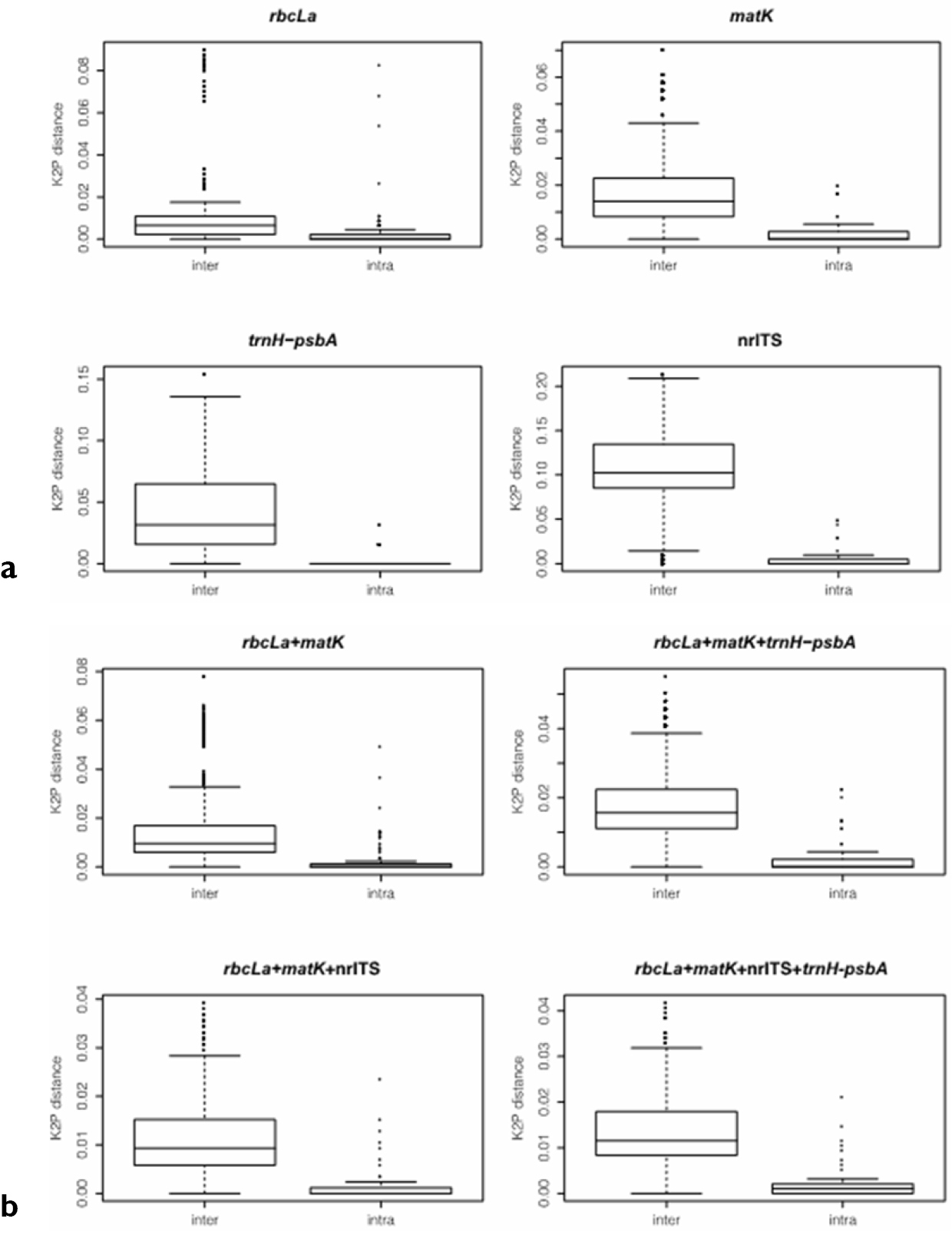
Comparisons of the distribution range of inter- versus intraspecific distances using boxplot a indicates comparison of single barcode gene regions b indicates the results of gene combinations.
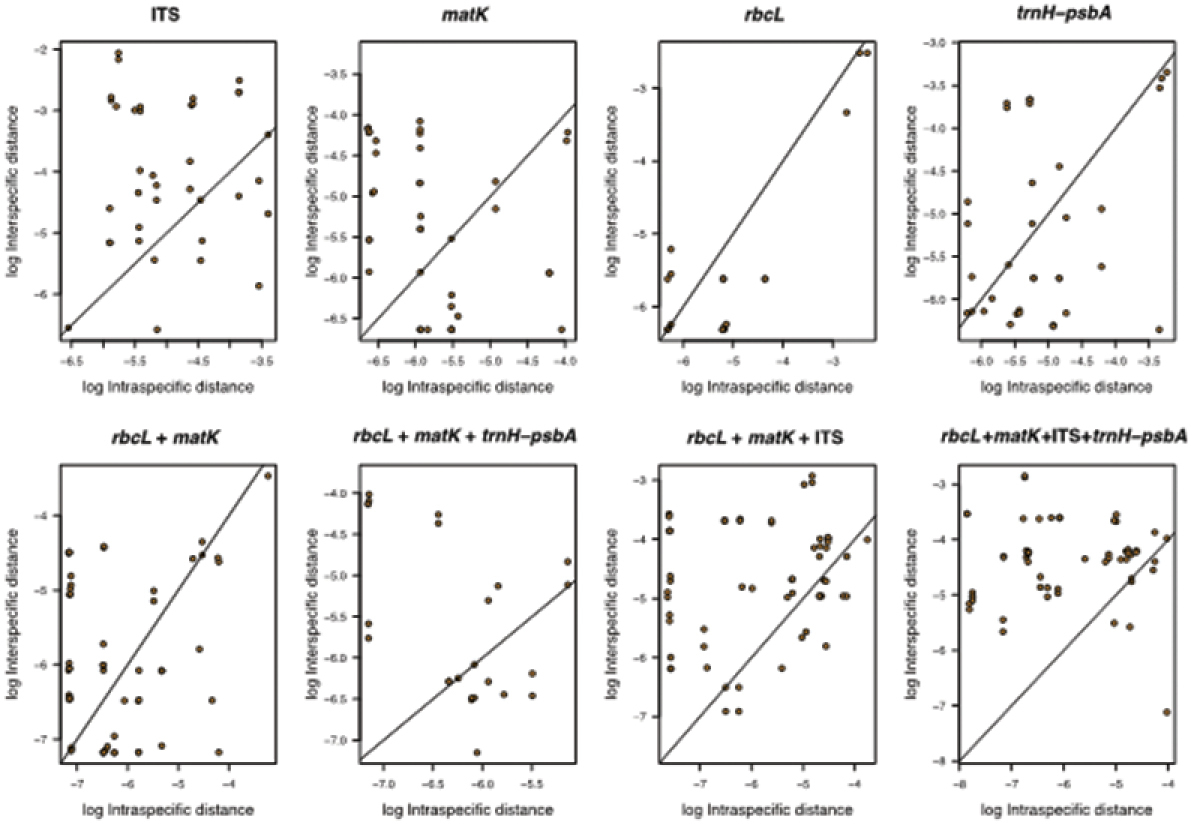
Relationships between inter- and intraspecific distances indicating barcoding gap for all regions tested.
Percentage barcode gap in all sequences for each region using the
| DNA region | Number of sequences without gap | Proportion of sequences with gap (%) |
|---|---|---|
| rbcLa | 132 | 13 |
| matK | 86 | 35 |
| trnH-psbA | 54 | 53 |
| nrITS | 25 | 73 |
| rbcLa+matK | 82 | 36 |
| rbcLa+matK+trnH-psbA | 37 | 57 |
| rbcLa+matK+nrITS | 27 | 64 |
| rbcLa+matK+nrITS+trnH-psbA | 11 | 84 |
Our results for the discriminatory power analysis varied with the methods applied (Table 3) at family level. Based on the Near Neighbour method, nrITS provided the highest discriminatory power (65%) followed by rbcLa + matK + trnH-psbA + nrITS (64%), rbcLa + matK + trnH-psbA (62%), and rbcLa + matK (61%). The lowest discriminatory power was found for trnH-psbA (28%).
Identification efficacy of DNA barcodes using distance based methods. F = False and T = True.
| DNA region | Near Neighbour | BOLD (1%) | Best Close Match | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F (%) | T (%) | Ambiguous (%) | Correct (%) | Incorrect (%) | No ID (%) | Ambiguous (%) | Correct (%) | Incorrect (%) | No ID (%) | |
| rbcLa | 59 | 41 | 61 | 18 | 14 | 7 | 61 | 18 | 14 | 7 |
| matK | 46 | 54 | 81 | 11 | 7 | 1 | 47 | 38 | 14 | 1 |
| trnH-psbA | 72 | 28 | 65 | 22 | 10 | 3 | 18 | 60 | 18 | 4 |
| nrITS | 35 | 65 | 29 | 47 | 10 | 14 | 10 | 63 | 19 | 8 |
| rbcLa+matK | 39 | 61 | 86 | 10 | 2 | 2 | 35 | 51 | 12 | 2 |
| rbcLa+matK+trnH-psbA | 38 | 62 | 79 | 16 | 2 | 3 | 6 | 80 | 8 | 6 |
| rbcLa+matK+nrITS | 43 | 57 | 62 | 30 | 7 | 1 | 3 | 70 | 19 | 8 |
| rbcLa+matK+nrITS+trnH-psbA | 36 | 64 | 52 | 41 | 3 | 4 | 0 | 87 | 9 | 4 |
BOLD species delimitation criteria of 1% threshold provided the lowest rate of correct identification among all three methods used. However, we found that nrITS remains the most efficient region with 47% discriminatory power. The second most successful combination of regions were core + trnH-psbA + nrITS (41%) followed by core + nrITS (30%) and trnH-psbA (22%); the core barcodes were identified as the least performing regions (10%) with the highest proportion of ambiguity (86%).
In contrast to the two previous methods, the Best Close Match provided the highest rate of species discrimination for the combined dataset (core + trnH-psbA + nrITS) yielding the best discriminatory power (87%) with no ambiguity. This was followed by core + trnH-psbA (80%), core + nrITS (70%) and nrITS (63%), with the poorest performance for rbcLa (18%) at family level.
The last criterion used to evaluate the potential of DNA region was PCR efficiency. We found that rbcLa (87%) followed by trnH-psbA (85%) and matK (68%) were easy to amplify, with nrITS being the most difficult (47%; Figure 5).
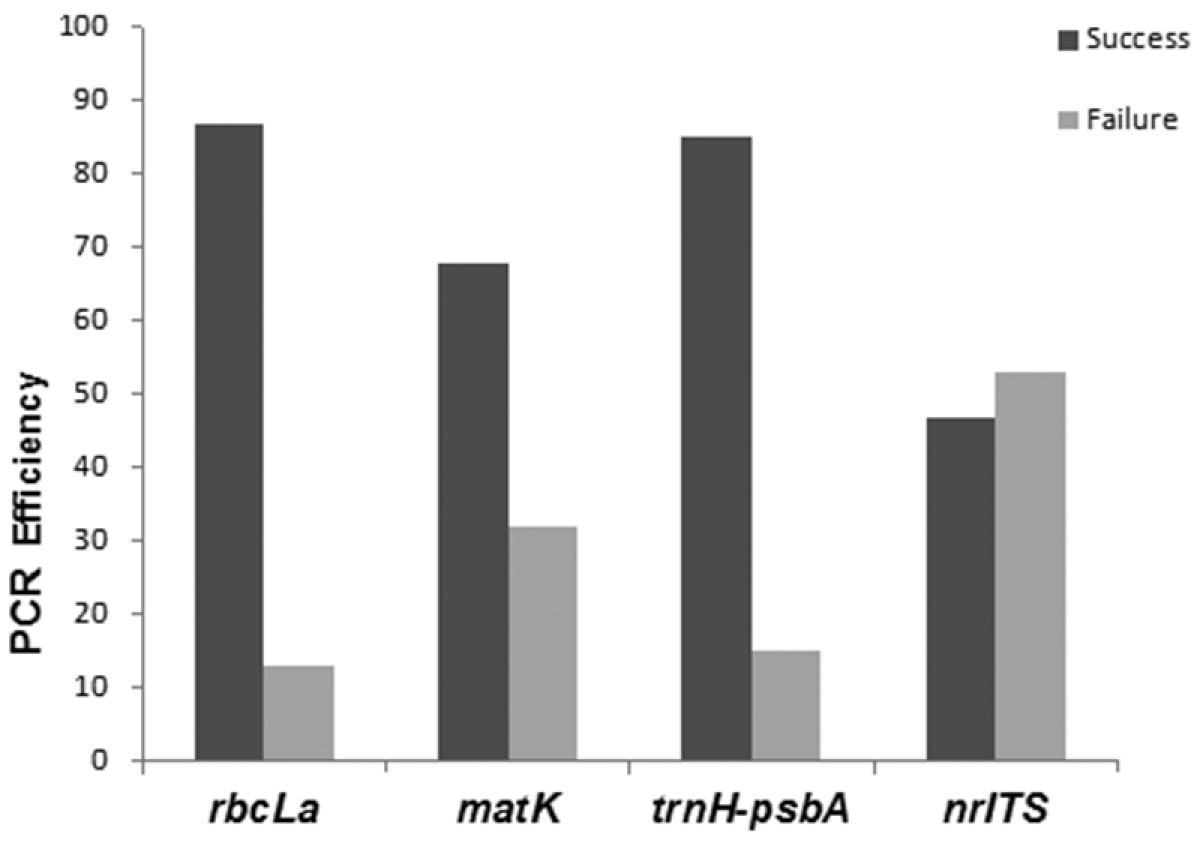
PCR efficiency for the four candidate barcodes (rbcLa, matK, trnH-psbA, nrITS).
We complemented previous analyses using species monophyly criteria after verifying the monophyly of Combretaceae. Among all regions, core + trnH-psbA isolated the highest proportion of monophyletic species (83%), followed by trnH-psbA (78%), nrITS (76%), and combination of all four regions (65%). Again, rbcLa provided the lowest performance in identifying species as monophyletic (37%; Figure 6).
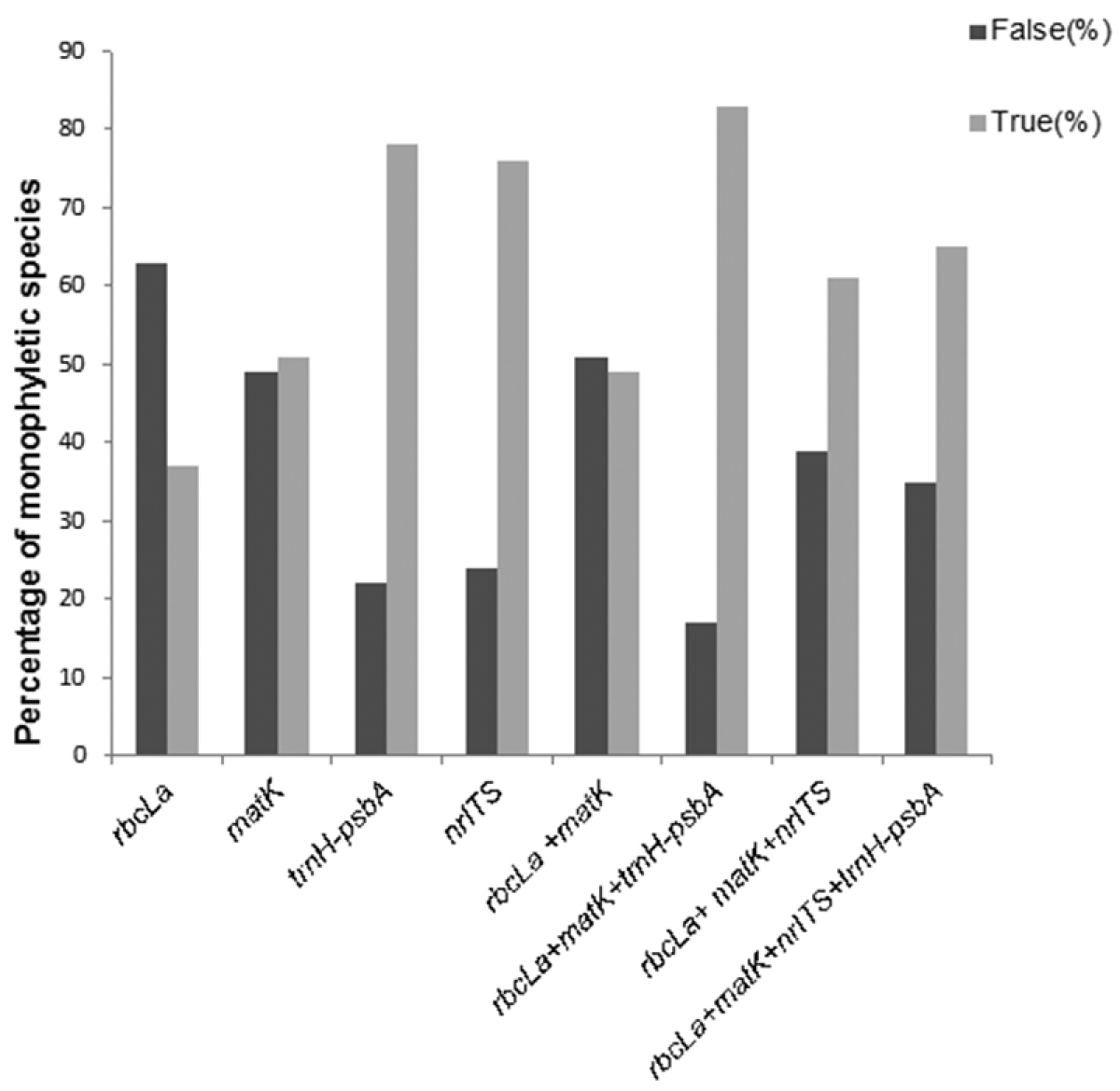
Gene performance based on monophyly criteria. False = proportion of non-monophyletic species; True = proportion of monophyletic species.
In summary, all regions provided evidence for barcode gaps (Figure 3a, b and 4), but the strength of evidence varied with approaches used. Furthermore, the Best Close Match method provided the highest identification accuracy among the three distance-based methods used irrespective of genes or combinations tested. Under this method, the two best potential barcodes for southern African Combretaceae were first, core+ trnH-psbA and second, core + trnH-psbA + nrITS. However, based on species monophyly criteria, the single region trnH-psbA and the combination core + trnH-psbA showed high barcode potential, with trnH-psbA being the second best easy-to-amplify region after rbcLa.
We further evaluated the potential of each region as candidate barcode using a phylogeny of southern African Combretaceae (Appendix 2). Our results are congruent to the corresponding subset in the most recent and largest phylogeny assembled for the family (Appendix 3). Our evaluation for the discriminatory power at subgeneric level using the thresholds determined for the family (1.31% for the core and 0.5% for the core + trnH-psbA) revealed that the core barcodes alone were able to correctly identify 78% of species within the subgenus Cacoucia. However, the core barcodes could discriminate only 50% of species within the subgenus Combretum. In particular, the discriminatory power of the core barcodes within both subgenera increased markedly to 100% when we added the trnH-psbA region (Table 4). This trend was consistent even when we applied the thresholds that have been optimised for the subgenera.
At sectional level, we observed similar trends – the addition of trnH-psbA increased the performances of the core barcodes drastically except for Macrostigmatea (Table 5): Angustimarginata (core: 11%; core + trnH-psbA: 86%); Ciliatipetala (core: 55%; core + trnH-psbA: 73%); Conniventia (core: 38%; core + trnH-psbA: 88%); Hypocrateropsis (core: 63%; core + trnH-psbA: 80%). However, Macrostigmatea (core 34%, core + trnH-psbA 44%) showed the least performance, even with the addition of trnH-psbA to the core barcode, with just 10% increment being observed. This trend is not sensitive to the thresholds applied for the family or the sections.
Comparisons of efficacy of core barcodes and best barcode within subgenera Combretum and Cacoucia.
| Subgenus | DNA region | No. of seq | Mean Inter (±SD) | Threshold (%) | Best Close Match | |||
|---|---|---|---|---|---|---|---|---|
| Ambiguous (%) | Correct (%) | Incorrect (%) | No ID (%) | |||||
| Cacoucia | rbcLa+matK | 23 | 0.004±0.002 | 1.31 | 13 | 78 | 9 | 0 |
| rbcLa+matK+trnH-psbA | 16 | 0.006±0.002 | 0.5 | 0 | 100 | 0 | 0 | |
| Combretum | rbcLa+matK | 84 | 0.009±0.009 | 1.31 | 36 | 50 | 12 | 2 |
| rbcLa+matK+trnH-psbA | 16 | 0.006±0.002 | 0.5 | 0 | 100 | 0 | 0 | |
Comparisons of core barcodes and the best barcode within five sections of the subgenera Combretum and Cacoucia.
| Sections | DNA regions | No. of seq | Mean inter (±SD) | Threshold (%) | Best Close Match | |||
|---|---|---|---|---|---|---|---|---|
| Ambiguous (%) | Correct (%) | Incorrect (%) | No ID (%) | |||||
| Angustimarginata | rbcLa+matK | 19 | 0.007±0.014 | 2.6 | 58 | 11 | 26 | 5 |
| rbcLa+matK+trnH-psbA | 15 | 0.006±0.006 | 0.7 | 0 | 86 | 7 | 7 | |
| Ciliatipetala | rbcLa+matK | 20 | 0.004±0.002 | 0.3 | 45 | 55 | 0 | 0 |
| rbcLa+matK+trnH-psbA | 15 | 0.006±0.003 | 0.5 | 0 | 73 | 27 | 0 | |
| Conniventia | rbcLa+matK | 8 | 0.005±0.004 | 0.8 | 37 | 38 | 12 | 13 |
| rbcLa+matK+trnH-psbA | 8 | 0.010±0.006 | 2.4 | 0 | 88 | 12 | 0 | |
| Hypocrateropsis | rbcLa+matK | 8 | 0.012±0.005 | 1.31 | 25 | 63 | 12 | 0 |
| rbcLa+matK+trnH-psbA | 5 | 0.020±0.004 | 0.8 | 0 | 80 | 20 | 0 | |
| Macrostigmatea | rbcLa+matK | 15 | 0.002±0.001 | 0.1 | 53 | 34 | 13 | 0 |
| rbcLa+matK+trnH-psbA | 9 | 0.003±0.002 | 0.2 | 0 | 44 | 56 | 0 | |
(Only sections with at least three different species are included).
Finally, we compared the mean number of substitutions between any two species within each section. We found that the mean number of substitutions between representatives of Macrostigmatea is lowest (mean = 4) whereas it ranges between 5 and 19 substitutions in other sections of subgenus Combretum.
We evaluated genetic variation for both single and various combinations of rbcLa, matK, trnH-psbA and nrITS. Comparing ranges of intra- versus interspecific distances, our results indicate that all markers show a barcode gap (
The discriminatory power of the DNA regions in species identification also varies with the distance-based methods applied. From the methods tested, Near Neighbour and Best Close Match yielded high performance, with the latter giving the best results for the possible three and four different gene combinations. The core barcodes were not recognised among the three best options, and its discriminatory power has been questioned in a number of studies (
However, our study indicates some important drawbacks that discount the inclusion of nrITS as a good barcode. For example, based on amplification success criteria, nrITS was the most difficult of all regions tested with rbcLa and trnH-psbA being the easiest regions to amplify. The technical hurdles in PCR amplification and sequencing of nrITS may be linked to the presence of retro-transposons and other repetitive elements within plant nuclear genomes, resulting in paralogous gene copies (
Previous studies have shown that core barcodes are very limited in discriminating taxa that are phylogenetically closely related, and suggested that the efficacy of DNA barcodes should be tested within a phylogenetic context (
While we found poor performance at sectional level, for example, in Angustimarginata, Macrostigmatea and Conniventia, this result is not unexpected due to a very low genetic variation one could expect within clades (see
The result for section Macrostigmatea reflects earlier tangle cited in previous studies regarding its composition (
Based on our results, the unclear taxonomy reported for section Macrostigmatea, is reflected, indicating a need for further molecular analyses involving more taxa and gene sequences to correctly determinemembers of this section. Our results also support the proposal of
Our analysis indicates that the poor performance of the core barcodes at family level could not be generalised to lower levels: the core barcodes perform poorly in some sections but shows strong discriminatory power in others. Such findings may indicate that the success of DNA barcodes in discriminating closely related species at least in plants may correlate with the evolutionary distinctiveness of the group tested and, as recently indicated, (see
We thank the Government of Canada through Genome Canada and the Ontario Genomics Institute (2008-OGI-ICI-03), The International Development Research Centre (IDRC), Canada and the University of Johannesburg for financial support and various local and international authorities granting us plant collections permits. We thank three anonymous reviewers for providing valuable comments on an earlier draft of the manuscript.
Supplementary table S1. (doi: 10.3897/zookeys.365.5728.app1) File format: Microsoft Excel file (xls).
Explanation note: Full names, voucher information, GenBank and BOLD accession numbers for taxa used in this study. A dash (—) indicates DNA regions not sampled and DNA sequences obtained from GenBank are underlined. Voucher specimens are deposited in the following herbaria: JRAU, University of Johannesburg (UJ), Johannesburg, South Africa; MO, Missouri Botanical Garden, St Louis, USA.
Supplementary figure S1. (doi: 10.3897/zookeys.365.5728.app2) File format: Microsoft Word file (docx).
Explanation note: One of most parsimonious trees obtained from the combined plastid and nuclear data (rbcLa, matK, trnH-psbA, and nrITS) set. Clades highlighted indicate the sections that were identified from the MP tree obtained from barcoding gene regions. Bootstrap percentages above 50% are shown above the branches.
Supplementary figure S2. (doi: 10.3897/zookeys.365.5728.app3) File format: Microsoft Word file (docx).
Explanation note: One of most parsimonious trees with branch tips collapsed from the combined plastid and nuclear data (rbcL, matK, psaA-ycf3, trnH-psbA, and nrITS) set. Clades highlighted indicate sections that were identified from the MP tree obtained from barcoding gene regions. Above the branches are Bayesian posterior probability (PP) values (> 0.5) and below are bootstrap percentages above 50%.