





(C) 2013 Zoltán T. Nagy. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
For reference, use of the paginated PDF or printed version of this article is recommended.
Citation: Nagy ZT, Sonet G, Mortelmans J, Vandewynkel C, Grootaert P (2013) Using DNA barcodes for assessing diversity in the family Hybotidae (Diptera, Empidoidea). In: Nagy ZT, Backeljau T, De Meyer M, Jordaens K (Eds) DNA barcoding: a practical tool for fundamental and applied biodiversity research. ZooKeys 365: 263–278. doi: 10.3897/zookeys.365.6070
Empidoidea is one of the largest extant lineages of flies, but phylogenetic relationships among species of this group are poorly investigated and global diversity remains scarcely assessed. In this context, one of the most enigmatic empidoid families is Hybotidae. Within the framework of a pilot study, we barcoded 339 specimens of Old World hybotids belonging to 164 species and 22 genera (plus two Empis as outgroups) and attempted to evaluate whether patterns of intra- and interspecific divergences match the current taxonomy. We used a large sampling of diverse Hybotidae. The material came from the Palaearctic (Belgium, France, Portugal and Russian Caucasus), the Afrotropic (Democratic Republic of the Congo) and the Oriental realms (Singapore and Thailand). Thereby, we optimized lab protocols for barcoding hybotids. Although DNA barcodes generally well distinguished recognized taxa, the study also revealed a number of unexpected phenomena: e.g., undescribed taxa found within morphologically very similar or identical specimens, especially when geographic distance was large; some morphologically distinct species showed no genetic divergence; or different pattern of intraspecific divergence between populations or closely related species. Using COI sequences and simple Neighbour-Joining tree reconstructions, the monophyly of many species- and genus-level taxa was well supported, but more inclusive taxonomical levels did not receive significant bootstrap support. We conclude that in hybotids DNA barcoding might be well used to identify species, when two main constraints are considered. First, incomplete barcoding libraries hinder efficient (correct) identification. Therefore, extra efforts are needed to increase the representation of hybotids in these databases. Second, the spatial scale of sampling has to be taken into account, and especially for widespread species or species complexes with unclear taxonomy, an integrative approach has to be used to clarify species boundaries and identities.
COI, cryptic species, DNA barcoding, geographic distances, taxonomy
With over 11 400 described species, the superfamily Empidoidea represents one of the largest extant lineages of flies (Diptera, Brachycera) (
Studies carried out over the last few decades indicate the family Hybotidae is to be monophyletic (
DNA barcoding (
In the current paper, we optimized protocols for DNA barcoding of hybotid flies and performed a preliminary barcoding study on selected genera and species of this group. We hope that this approach will accelerate an inventory of hybotid flies. Here, we investigated the ability of the barcoding data coming from a large sampling of diverse Hybotidae to reveal cryptic species, patterns of geographic variation, and putative new species.
A total of 339 specimens, representing 164 morphospecies of Hybotidae (see Supplementary file 1) were selected and sequenced for this study. All material was collected between 2008 and 2012 in three biogeographic realms: the Palaearctic (Belgium, Portugal, France and Russian Caucasus), Afrotropical (D. R. Congo) and Oriental (Singapore and Thailand) realms. Our outgroup taxon was Empis tessellata Fabricius, 1794 (Empidoidea, Empididae), of which two individuals were sequenced. Specimens were collected mainly using sweep nets and Malaise traps, and were initially preserved in 70% ethanol. After identification, all specimens were transferred to 96% ethanol and then stored at 4 °C in order to minimize DNA degradation. Either complete specimens or mid or hind legs were used for total genomic DNA extraction. Immediately prior to extraction, each tissue sample was placed in a microtube and air-dried. DNA extractions were carried out using the NucleoSpin Tissue kit (Macherey-Nagel). We followed the manufacturer’s instructions, but used a longer lysis time (i.e. around 24 hours). After lysis, fly specimens were transferred to absolute ethanol and were put back to the collection. Voucher specimens have been deposited in the entomological collections of the Royal Belgian Institute of Natural Sciences (RBINS).
PCR conditions were optimized by testing primer concentrations of 0.1, 0.2 and 0.4 µM and MgCl2 concentrations of 1.5–2 mM against a gradient of annealing temperatures. The best results were obtained by using the protocol as follows: each reaction (total volume of 25 µl) contained 2–3 µl DNA extract, 0.4 µM of each primer, 0.03 unit/µl Platinum Taq polymerase (Invitrogen), 1 × PCR Buffer, 0.2 mM dNTP, 2 mM MgCl2 and ca. 15 µl of sterile water. The COI region of interest was amplified using the standard animal barcoding primers, LCO1490 and HCO2198 (
DNA sequences were checked and assembled with SeqScape v2.5 (Life Technologies). Neighbour-Joining (NJ) trees based on uncorrected (p) distances were calculated in MEGA5 (
Our sampling covered all the currently accepted subfamilies and tribes of the Hybotidae. At the generic level, we investigated 22 of the 66 known genera (see Table 1 for full details). The DNA sequence data set consisted of 341 COI sequences (339 Hybotidae and two Empididae), each sequence being of 657 bp in length. These sequences were deposited in BOLD and GenBank (BOLD Process IDs EMPID001-13–EMPID341-13).
Global suprageneric systematics of Hybotidae (without the genus Stuckenbergomyia) and genera investigated in the current barcoding study.
| Subfamily (Tribe) | Number of genera | Investigated genera |
|---|---|---|
| Trichininae | 2 | 1 (Trichina) |
| Ocydromiinae | 15 | 3 (Leptopeza, Ocydromia, Oropezella) |
| Oedaleinae | 4 | 3 (Allanthalia, Euthyneura, Oedalea) |
| Tachydromiinae | ||
| - Symballophthalmini | 1 | 1 (Symballophthalmus) |
| - Tachydromiini | 8 | 4 (Ariasella, Platypalpus, Tachydromia, Tachypeza) |
| - Drapetini | 18 | 6 (Chersodromia, Crossopalpus, Drapetis, Elaphropeza, Nanodromia, Stilpon) |
| Hybotinae | ||
| - Bicellariini | 13 | 1 (Bicellaria) |
| - Hybotini | 14 | 3 (Hybos, Syndyas, Syneches) |
An NJ tree without species names and additional sample information is shown in Figure 1 (a fully annotated tree is shown in Supplementary file 2). ‘Species-level’ groups (i.e., close to or at terminal nodes) were generally well supported, while deep-level groups were not (see Figure 1 and Supplementary file 2). Especially when low intraspecific distance was observed, these groups (considered as molecular operational taxonomic units – MOTUs) often received 100% bootstrap support. At a 1% distance threshold (as it is also used by BOLD), 99% of the clusters (i.e., 70 out of 71 MOTUs) were supported by bootstrap values above 95%. Many recognized species were well resolved and distinguished using the COI data, but we observed a number of problems that are discussed below. Although representatives of more inclusive taxa, such as tribes and subfamilies (with the exception of the Ocydromiinae and Tachydromiini), were usually recovered in single clusters, these clusters were not supported (bootstrap values < 70%). The only exception is the tribe Symballophthalmini, represented in the data set by just two species, which was supported by a bootstrap value of 87.7%.
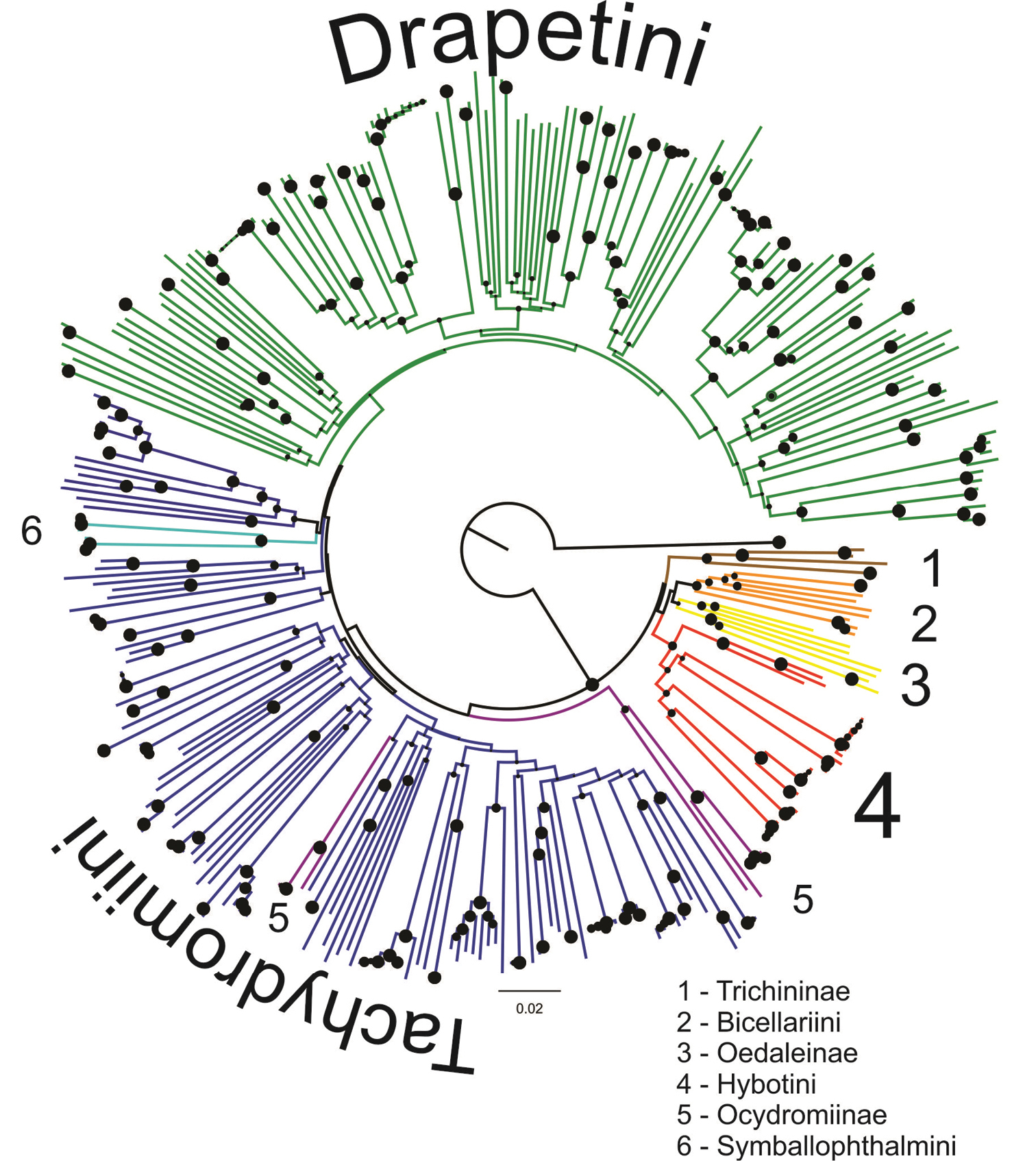
Neighbour-Joining tree representing hybotid diversity of 339 selected samples. The tree was rooted with Empis tessellata (Empididae). Circles represent branch supports, bootstrap values are according to circles’ size.
For most genera, the number of species represented in our analysis was very limited. Similarly, the number of conspecific sequences was also generally low, ranging between 1–9. Nonetheless, we observed considerable overlaps between intraspecific (0–17.2%) and interspecific divergences (0–21.81%). Among congeners, interspecific divergences ranged between 0–19.9%, while we observed higher divergence between samples of different genera (5.85–21.81%). Hence, no barcoding gaps existed between any of these ranks. The ranges of pairwise distances were overall high in the four genera represented by the highest number of samples (Table 2). We observed extensive overlap between intra- and interspecific divergences in both the species-rich genera of Tachydromiini, Platypalpus and Tachydromia, with less extensive, or no overlap, in the genera Chersodromia and Elaphropeza, both belonging to the Drapetini (Table 2).
Below, we describe five categories of cases where ranges of intra- and interspecific distances did not seem consistent with the current taxonomy and would require more investigation.
Patterns of intra- and interspecific distances observed in four species-rich genera of our dataset.
| Tribe | Genus | No. of species | No. of sequences | No. of haplotypes | Intraspecific distances (%) | Interspecific distances (%) |
|---|---|---|---|---|---|---|
| Tachydromiini | Platypalpus | 45 | 98 | 81 | 0–16.89 | 0–18.72 |
| Tachydromiini | Tachydromia | 12 | 21 | 18 | 0–5.48 | 1.07–18.11 |
| Drapetini | Chersodromia | 12 | 36 | 26 | 0–3.04 | 6.09–15.53 |
| Drapetini | Elaphropeza | 43 | 75 | 68 | 0–5.48 | 1.83–19.63 |
We found that in some congeneric species the levels of sequence variation observed both within populations and between populations in close proximity were low. Contrastingly, other congeneric species showed widely different levels of intraspecific divergence. An interesting case in this respect is the brachypterous Chersodromia curtipennis and the fully-winged Chersodromia pontica, both of which occur on the Taman Peninsula (Krasnodar region of Russia). Samples were taken at various sites on the Taman Peninsula, ranging from the North, on the coast of the Sea of Azov, to the South along the Black Sea (Taman: Veselovka). While the brachypterous species showed virtually no genetic variation (uncorrected pairwise divergence was between 0–0.15%), the fully-winged species showed an expressed pattern of divergence with pairwise p-distances of 0–1.37% (Figure 2A).
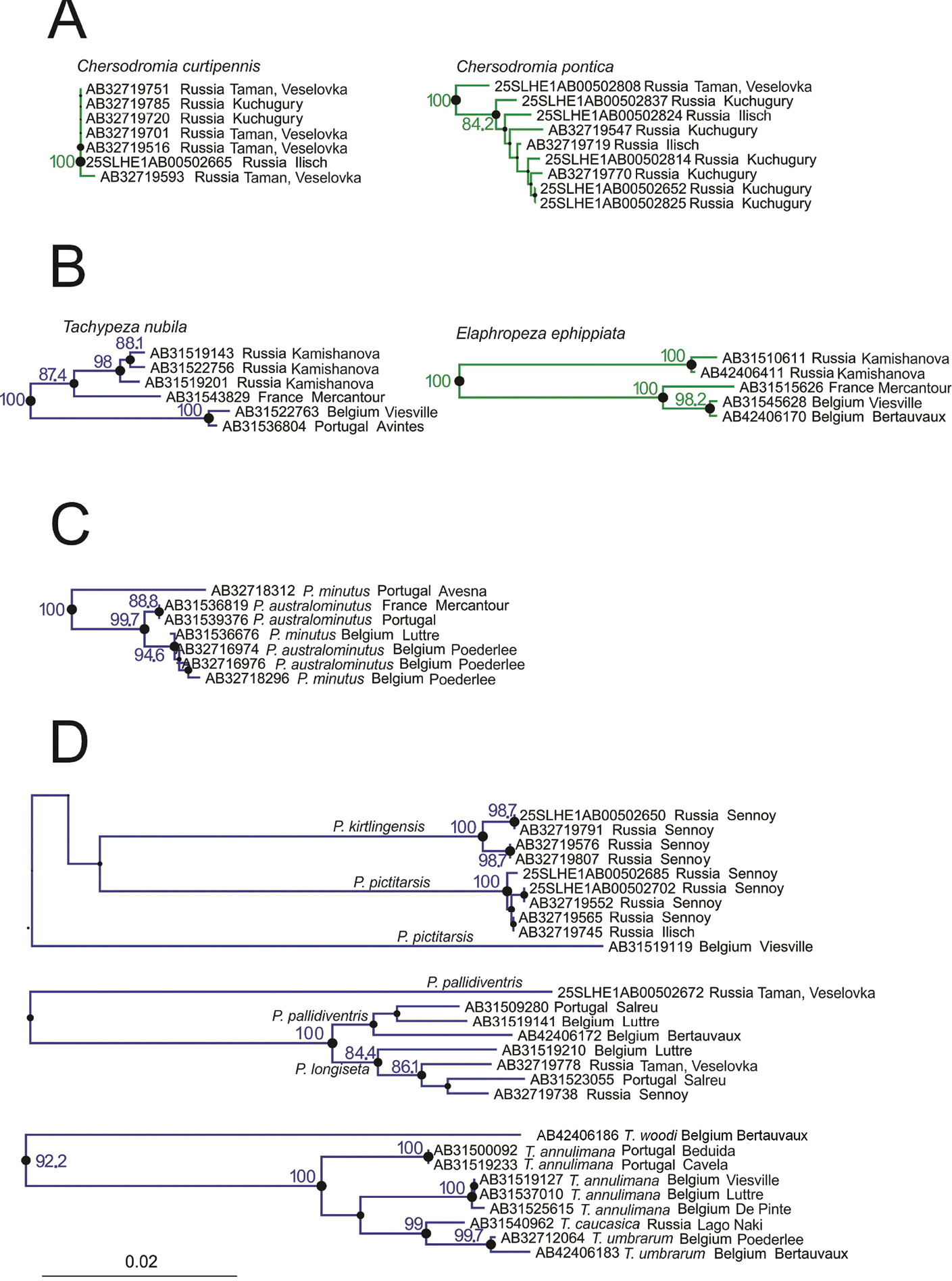
Subtrees showing cases where ranges of intra- and interspecific distances do not seem consistent with the current taxonomy and would require more investigation. See details in text. Circles represent branch supports. Bootstrap values are according to circles’ size, bootstrap values are shown in numbers when > 80%.
Some species sampled across large geographical areas showed high levels of genetic divergence between populations. This is among others the case for two species that are widespread and very common in Europe: Tachypeza nubila and Elaphropeza ephippiata. While we could not detect any morphological differences (i.e. of the body and the male genitalia) between populations in western European and the Russian Caucasus, intraspecific pairwise genetic divergences ranged between 0.3–3.5% in Tachypeza nubila and between 0.2–5.48% in Elaphropeza ephippiata (Figure 2B). Unexpectedly large ‘intraspecific’ divergences may indicate undescribed diversity at the species level. In many cases, large ‘intraspecific’ divergences were found between specimens from the same locality or from adjacent sites (Table 3, upper part), and examples in this respect include Platypalpus caucasicus (Russian Caucasus), Platypalpus annulipes (Belgium), Trichina elongata (Russian Caucasus), Bicellaria nigra (Russian Caucasus), Tachydromia annulimana (within Europe), Elaphropeza nuda (D. R. Congo) and Elaphropeza monospina (Singapore). In a number of other cases, large ‘intraspecific’ divergences were observed between geographically distant populations (Table 3, lower part); this was observed for Platypalpus pictitarsis (Russian Caucasus versus Belgium), Platypalpus pallidiventris (Russian Caucasus versus Europe), Leptopeza flavipes (Russian Caucasus versus Belgium), Oedalea zetterstedti (Russian Caucasus versus Belgium), Euthyneura myrtilli (Russian Caucasus versus Europe), Platypalpus nigritarsis (Russian Caucasus versus France) and Tachydromia aemula (Russian Caucasus versus Portugal). In all of these cases, morphological differences of genitalia (or other diagnostic characters) were not assessed in details, and therefore these divergences may well reflect interspecific differences. Remarkably, no significant differences in divergence ranges were observed between the two types of cases (i.e., associated or not with large spatial distances).
Range of pairwise p-distances in cases where unexpectedly high ‘intraspecific’ divergence was observed (> 5%).
| Species or species complex | Range of pairwise p-distances (%) |
|---|---|
| Platypalpus caucasicus | 0.46–8.07 |
| Platypalpus annulipes | 0–9.80 |
| Trichina elongata | 0.91–8.83 |
| Bicellaria nigra | 9.44 |
| Tachydromia annulimana | 0–10.35 |
| Elaphropeza nuda | 0–5.33 |
| Elaphropeza monospina | 5.33 |
| Platypalpus pictitarsis | 0–10.20 |
| Platypalpus pallidiventris | 1.37–10.05 |
| Leptopeza flavipes | 0–7.01 |
| Oedalea zetterstedti | 7.91 |
| Euthyneura myrtilli | 1.52–10.96 |
| Platypalpus nigritarsis | 5.33 |
| Tachydromia aemula | 5.48 |
Platypalpus minutus and Platypalpus australominutus are externally very similar except that male genitalia are consistently different (
In Figure 2D, three examples are shown where the unclear taxonomy of the involved species or species complex was reflected in para- or polyphyletic taxa. For example, Platypalpus pictitarsis and Platypalpus kirtlingensis are morphologically very similar. They differ in the colour of the fore leg and the palpus. Barcode sequences showed that both species are genetically different (uncorrected pairwise interspecific divergence was at least 8.37%). In addition, however, a single male of Platypalpus pictitarsis from Belgium (AB31519119) rendered this taxon polyphyletic. Both external morphology and genitalia of the Belgian and Caucasian pictitarsis was the same and a deeper study will be needed to clarify this issue.
Another example involved the sister species Platypalpus pallidiventris and Platypalpus longiseta, which differ morphologically only in a few but distinct characters. Also, these species were genetically closely related except a single specimen of Platypalpus pallidiventris (25SLHE1AB00502672, see Figure 2D), which rendered this species paraphyletic. This single specimen of Platypalpus pallidiventris from Caucasus is a female, exhibiting less diagnostic characters than males, and could therefore belong to another species. This observation urges for a more intensive collection and study of these sister species. When we discarded this divergent sequence, both species showed a moderate intraspecific structuring. The bootstrap value supporting the cluster containing both species without the divergent specimen was 100%. In addition, the reciprocal monophyly of both species was supported with bootstrap values of 77.3% and 84.4% for Platypalpus pallidiventris and Platypalpus longiseta, respectively.
A third example involved four species (Figure 2D). Originally, a female (AB42406186) of Tachydromia woodi was identified as Tachydromia annulimana. However, the considerable divergence at COI between this specimen and all other specimens of Tachydromia annulimana (10.35%) suggested a misidentification. A reexamination of the specimen revealed that Tachydromia woodi has the costa between vein R1 and R2+3 thickened, an unpublished feature that confirmed the misidentification. Tachydromia caucasica from Caucasus and Tachydromia umbrarum from Belgium, both also belonging to the annulimana-group, showed a very low interspecific divergence (1.07-1.52%). This suggests, in combination with the little morphological differences reported between the two species, a very close relationship between the two species and does not exclude that they are conspecific.
The barcoding of dipterans commenced relatively early as part of the global DNA barcoding initiative. On the one hand, DNA barcoding performed well in several lineages. However, so far mostly Holarctic dipterans have been investigated where species diversity is overall lower than in tropical biomes. For instance, DNA barcoding proved to work well for Canadian (
In the meantime, several dipteran barcoding projects have been started, particularly with respect to medically, forensically or commercially (e.g., related to agriculture) relevant lineages such as mosquitoes, muscids, tephritids and drosophilids (see details at http://boldsystems.org). Hybotids, or more broadly the empidoids, have no known medical, forensic or commercial importance, therefore there are overall much less intensively studied. The current dataset presented herein is a result of a pilot study focusing on Old World hybotid diversity. An overall high sequence divergence was observed in our dataset, which is not surprising in the light of the age and diversification pattern of dance flies (
Our limited sampling of specific and subspecific levels with up to nine samples per species did not allow us to perform extensive tests on barcoding performance and species (or genus) delimitations. We are also aware of the potential pitfalls when analyzing taxonomically incomplete datasets. In such cases, a hierarchical sampling should be followed whenever possible. In these cases, the number of sampled genera and more inclusive taxa should be maximized (
In the case of recently diverged species, a number of methods have been compared (
Although we focused on problematic or unexpected cases, in most of these examples, DNA barcoding may still be useful, provided that precautions are taken with respect to taxonomic and geographic sampling effects. Moreover, species identification in Hybotidae is based primarily on male terminalia and possibly some of the species concept situations are due to misidentification of females. Also, collecting precise information on collection site, life history, habitat, morphology etc. can very well contribute to the interpretation of the DNA barcoding results. Our finding about intraspecific divergence patterns in the brachypterous vs. the fully-winged species (Chersodromia curtipennis and Chersodromia pontica, respectively) exemplifies this well. The reduced mobility of the brachypterous species is apparently linked to the low intraspecific diversity but mechanisms are still unclear. In many cases where we found unexpectedly large intraspecific divergences (Table 3), we probably deal with undescribed species, and therefore, in fact, with interspecific divergences. Nevertheless, further investigations are necessary to clarify the taxonomic status of the divergent populations. We advocate in-depth investigations involving more diagnostic traits and multi-gene analyses, evaluated in an integrative taxonomic framework (
In the current study, we provided a baseline for further studies on hybotid diversity using a DNA barcoding approach. We provided an optimized lab protocol for routine barcoding. We conclude that DNA barcoding can assist to identify hybotid taxa. Also cryptic species may be revealed by appropriate genetic markers, mostly because the morphological differences are not well assessed. Nevertheless, we emphasize to have an integrative look on barcoding data, and use this approach as a pragmatic first step in taxonomic practice or for biodiversity assessments.
We thank Jean-Luc Renneson, Marc Pollet, Pol Limbourg, Alain Drumont (Belgium), Christophe Daugeron (France), Rui Andrade (Portugal), Igor Shamshev and Semen Kustov (Russia) and Adrian Plant (United Kingdom) for their assistance. We also thank three anonymous reviewers for their constructive comments. We appreciate the assistance of Dinarzarde Raheem (Belgium), her comments and corrections improved the text significantly. We also thank the National Parks of Singapore (Singapore) and the University of Kisangani (D. R. Congo). JEMU is financed by the Belgian Science Policy Office (Belspo).
Samples used in the current study. (doi: 10.3897/zookeys.365.6070.app1) File format: Microsoft Excel file (xls).
Neighbour-Joining tree representing hybotid diversity of 339 selected samples. (doi: 10.3897/zookeys.365.6070.app2) File format: Adobe PDF (pdf).
Explanation note: Neighbour-Joining tree representing hybotid diversity of 339 selected samples. The tree was rooted with Empis tessellata (Empididae). Bootstrap support was estimated with 1000 replicates.