





(C) 2013 Herman Van Der Bank. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
For reference, use of the paginated PDF or printed version of this article is recommended.
Citation: Van Der Bank HF, Herbert D, Greenfield R, Yessoufou K (2013) Revisiting species delimitation within the genus Oxystele using DNA barcoding approach. In: Nagy ZT, Backeljau T, De Meyer M, Jordaens K (Eds) DNA barcoding: a practical tool for fundamental and applied biodiversity research. ZooKeys 365: 337–354. doi: 10.3897/zookeys.365.5356
The genus Oxystele, a member of the highly diverse marine gastropod superfamily Trochoidea, is endemic to southern Africa. Members of the genus include some of the most abundant molluscs on southern African shores and are important components of littoral biodiversity in rocky intertidal habitats. Species delimitation within the genus is still controversial, especially regarding the complex O. impervia / O. variegata. Here, we assessed species boundaries within the genus using DNA barcoding and phylogenetic tree reconstruction. We analysed 56 specimens using the mitochondrial gene COI. Our analysis delimits five molecular operational taxonomic units (MOTUs), and distinguishes O. impervia from O. variegata. However, we reveal important discrepancies between MOTUs and morphology-based species identification and discuss alternative hypotheses that can account for this. Finally, we indicate the need for future study that includes additional genes, and the combination of both morphology and genetic techniques (e.g. AFLP or microsatellites) to get deeper insight into species delimitation within the genus.
Mollusca, Gastropoda, Trochidae, species delimitation, morphology
Molluscs comprise one of the largest marine phyla, comprising more than 50 000 described species (marine species only), of which less than 10% are currently included in the global database of DNA barcodes (
Although the mitochondrial cytochrome c oxidase I gene (COI), used for barcoding purposes of animals is not efficient for all taxonomic groups (e.g. terrestrial gastropods,
In this study, our main objective was to infer species boundaries within the genus using DNA barcode. To date, attempts to resolve taxonomic issues within the genus using DNA sequence data were very limited in sample size: only one individual of each of the five recognised Oxystele species was generally analysed. For this purpose, we sampled 56 specimens including all five Oxystele species from a wide geographic distribution range. We then applied the DNA barcoding approach for taxa delimitation.
Sampling sites were widely distributed to cover the geographical distribution range of the genus. Species identification was done using the morphological characters given in the key to Oxystele species provided by
DNA extraction, polymerase chain reactions (PCR) and sequencing of the COI region (animal DNA barcode) were done at the Canadian Centre for DNA Barcoding (CCDB). PCR reactions followed standard CCDB protocols as described by
Species, authority, GenBank accession numbers (DQ) and BOLD process ID numbers (HVDBM) of specimens studied. Specimens in bold are those for which morphological characters (weathered shell colours and patterns) failed to provide accurate identification; this is revealed in the barcoding test of species delimitation and in phylogenetic tree topology. Sample localities for Oxystele impervia and Oxystele variegata individuals are indicated: southern Cape1, Robben Island2, north-western Cape3, Namibia4
We assessed the “DNA barcode gap” (
We also tested the discriminatory power of DNA barcoding by evaluating the proportion of correct species identification using the COI region. All sequences were labeled according to the names of the species from which the sequences were generated. The test of discriminatory power works as follows. Each sequence is considered as an unknown while the remaining sequences in the dataset are considered as the DNA barcode database used for identification. If the identification of the query is the same as the pre-considered identification (i.e. the sequence labels), the identification test is scored as “correct”, and the overall proportion of correct identification corresponds to the discriminatory power of the region tested, i.e. COI. This test was done applying three approaches: the “best close match” (
The function bestCloseMatch conducts the “best close match” analysis of
The function threshID conducts a threshold-based analysis based on a threshold genetic distance of 1% as conducted by the “Identify Specimen” tool provided by the BOLD system (http://www.boldsystems.org/views/idrequest.php). It is more inclusive than bestCloseMatch, in that it considers all sequences within the threshold of 1%. There also four possible outcomes for threshID tests, that is, “correct”, “incorrect”, “ambiguous”, and “no id” similar to the outcomes of the bestCloseMatch function.
The nearNeighbour function finds the closest individual and returns the score “true” (equivalent to “correct”) if their names are the same, but if the names are different, the outcome is scored as “false” (equivalent to “incorrect”).
Further, we performed a barcoding test of taxon delimitation. In reality, this test groups specimens into “molecular operational taxonomic units” (MOTUs;
Finally we complemented the test of MOTU delimitation with a phylogenetic analysis of COI sequences. We reconstructed a phylogenetic tree using Bayesian and maximum parsimony methods. The Bayesian tree was reconstructed using MrBayes v3.1.2 (
Maximum parsimony (MP) was implemented to analyse the data using PAUP* v4.0b10 (
Jujubinus exasperatus (Pennant, 1777) was used as outgroup based on
Our dataset includes 56 specimens: nine specimens of Oxystele impervia, 12 of Oxystele sinensis, four of Oxystele tabularis, 11 of Oxystele tigrina, and 20 specimens of Oxystele variegata (Table 1). The aligned COI matrix was 654 base pairs in length, including A: 24.2%; C: 21.1%; G: 18.3% and T: 36.4%.
Interspecific distances range from 0 to 0.18 (median = 0.15) and are generally larger than intraspecific distances (range: 0-0.09; median = 0.004; Wilcoxon test, p < 0.001; Figure 1A). This indicates that there is a barcode gap in the dataset. Even when we compared the lowest interspecific versus the furthest intraspecific distance, we also found that barcode gap exists within the COI sequences (grey lines in Figure 1B).
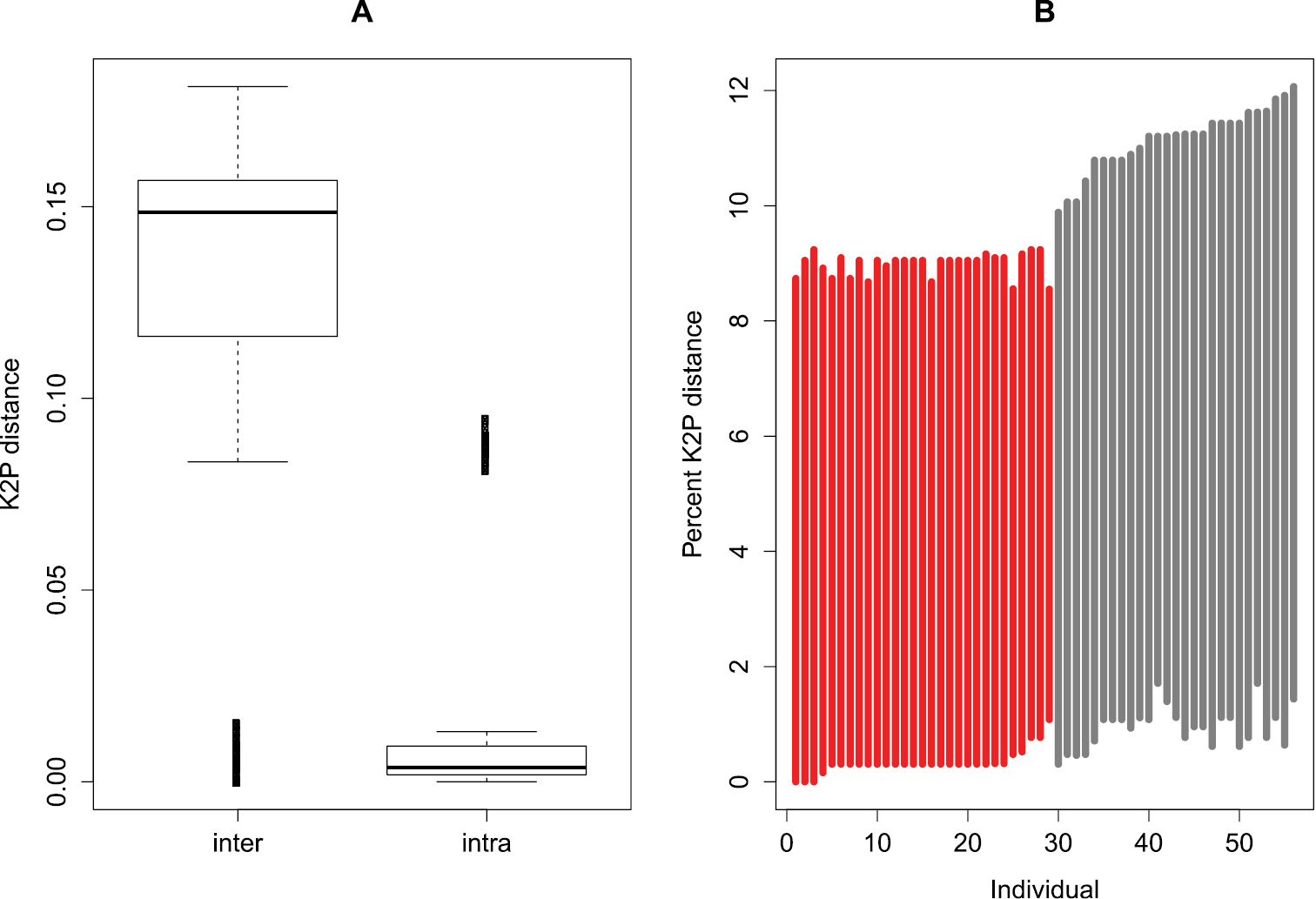
Evaluation of barcode gap in the dataset. A Boxplot of the interspecific (inter) and intraspecific genetic (intra) distances, indicating the existence of a barcode gap i.e. intraspecific distance is longer than intraspecific distance. The bottom and top of the boxes show the first and third quartiles respectively, the median is indicated by the horizontal line, the range of the data by the vertical dashed line and outliers (points outside 1.5 times the interquartile range) by Bold vertical lines B Lineplot of the barcode gap for the 56 Oxsystele specimens. For each specimen in the dataset, the grey lines indicate where the smallest interspecific distance (top of line value) is longer than the longest intraspecific distance (bottom of line value), therefore indicating existence of barcode gap; the red lines show where this pattern is reversed, and the closest non-conspecific is closer to the query than its nearest conspecific, i.e., the situation where there is no barcoding gap.
We determined the optimised threshold genetic distance (d) with which we tested the discriminatory power of COI sequences and delimited MOTUs. We found d = 0.047 (Figure 2). Testing the efficacy of DNA barcoding based on this threshold, we found that COI sequences performed very well in assigning DNA sequences to the correct species (Table 2). For instance, under both near neighbour and best close match methods, 87.5% of the COI sequences were correctly identified (49 specimens out of 56). However, the best close match method indicates 5.36% of ambiguity (three specimens), i.e. both correct and incorrect species are within the given threshold; and 7.14% of incorrect identification (four specimens). Also, for 12.5% of sequences (seven specimens) the near neighbour method results in “incorrect”. Using the BOLD method (threshold = 1%), we obtained poor barcoding performance, that is, we have as many correct as ambiguous results (48.21% respectively; i.e. 27 specimens). The BOLD method also indicates one “incorrect” and one “no id” (Table 2).
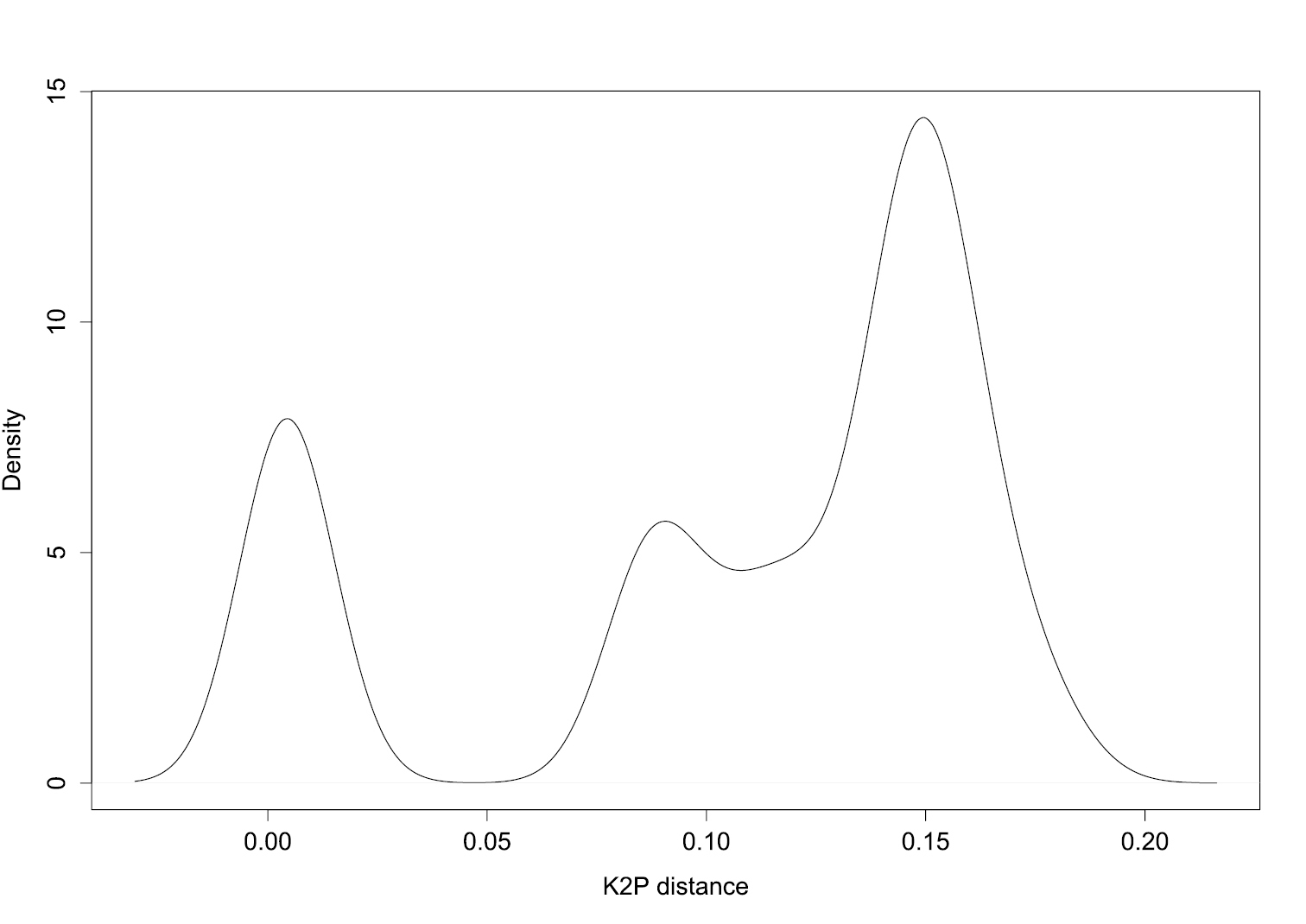
Determination of the threshold genetic distance for species identification. The density plot indicates transition between intra- and interspecific distances; the genetic distance corresponding to this transition (dip in the density graph, here approximately 0.05) indicates the suitable threshold to the dataset. This method does not require prior knowledge of species identity to get an indication of potential threshold values.
Tests of barcoding identification accuracy with numbers (n) and percentages (%) of each score.
| Methods | Near neighbour | Best Close match | BOLD criteria | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Scores | False | True | Ambiguous | Correct | Incorrect | No ID | Ambiguous | Correct | Incorrect | No ID |
| n (%) | 7 (12.5%) | 49 (87.5%) | 3 (5.36%) | 49 (87.5%) | 4 (7.14%) | 0 (0%) | 27 (48.21%) | 27 (48.21%) | 1 (1.79%) | 1 (1.79%) |
Further, all the 56 specimens included in this study were grouped into five MOTUs based on our threshold (Table 1). Using tree-based analysis, we also found five strongly supported groupings (PP = 1.00; BP = 100%), identified as A–E (Figure 3), except that the grouping B corresponding to Oxystele variegata is only well supported in the MP analysis (BP = 98%). The composition of these five groupings matches that of MOTUs and comprises Oxystele tabularis (A), Oxystele variegata (B), Oxystele impervia (C), Oxystele sinensis (D), and Oxystele tigrina (E) (Figures 3, Appendices 1 and 2).
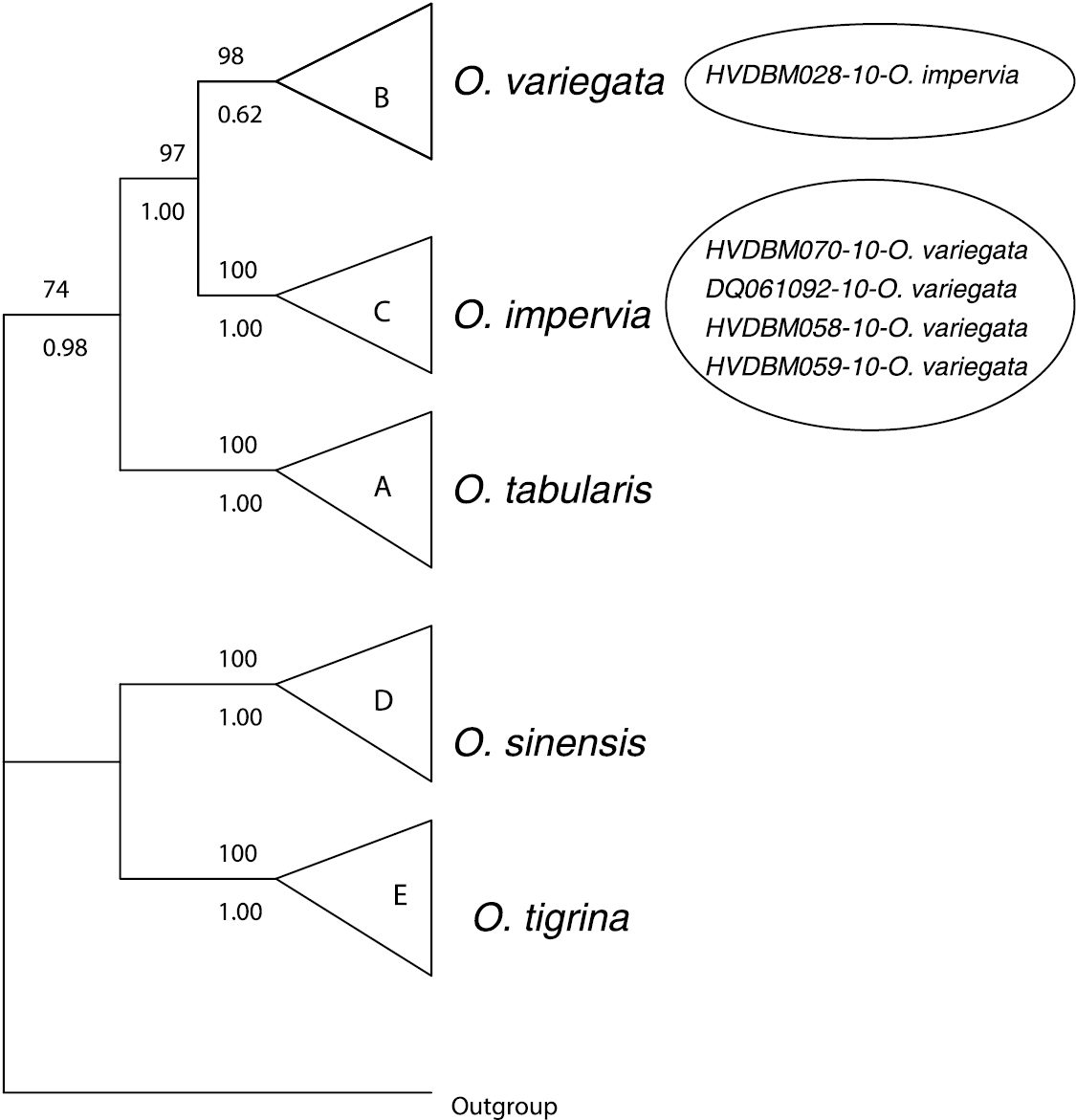
Summary of both Bayesian and parsimonious trees. Values above branches indicate bootstrap supports; values under branches indicate posterior probability. All distinguished species are indicated at the tip of the tree. Branches without values indicate non-supported nodes; the small circle indicates a specimen of Oxystele impervia (HVDBM028-10) that was misidentified based on morphology; large circle indicates four specimens morphologically indistinguishable from Oxystele variegata (HVDBM070-10; DQ061092; HVDBM058-10; HVDBM059-10), but that are, based on both barcoding analysis of species delimitation (see Table 1) and phylogenetic tree analysis identified as Oxystele impervia (see also Appendices 1 and 2).
The concept of DNA barcoding was first proposed as a technique to accelerate species identification within micro-organisms (
We first tested COI’s potential as a good barcode for the genus Oxystele. A good barcode candidate is expected to exhibit a barcode gap (
In addition, we found that COI has a strong discriminatory power (85%) within the genus Oxystele especially using the best close match and near neighbour methods. This gives support to the efficacy of COI for identification purposes within the genus. However, the application of BOLD identification criteria yields a poor identification success i.e. < 50% and similar proportion of ambiguity (Table 2). The poor performance of COI using BOLD criteria should not be seen as a result of barcoding inefficiency, but should rather be linked to the untested 1% threshold used in BOLD identification (see
Our analysis of barcoding-based taxon delimitation results in five MOTUs, of which three correspond to morphology-delimited species: Oxystele sinensis, Oxystele tabularis and Oxystele tigrina (Table 1).These results are also supported by phylogeny-based analysis of species delimitation. However, four specimens identified morphologically as Oxystele variegata are included by the barcoding taxon delimitation test within the MOTU of Oxystele impervia. Similarly, one specimen identified morphologically as Oxystele impervia is grouped within the MOTU of Oxystele variegata (Figure 3). These mismatches between morpho-species identification and barcoding-based taxon delimitation (MOTUs) reflect the controversy surrounding species boundaries and/or the identification key (e.g.
Why the mismatch between MOTU and morpho-species? Potential explanations include unsuitable morphology-based taxon delimitation, species paraphyly (– including but not restricted to ancestral polymorphism), and on-going gene flow (i.e., the two taxa are not distinct species or they hybridize; see
In our attempt to resolve the taxonomic uncertainty, we also used the phylogenetic tree reconstruction. The results are similar to those of MOTUs, that is, one specimen morphologically identified as Oxystele impervia, grouped on the phylogeny with Oxystele variegata (grouping B, Figure 3, Appendices 1 and 2), but this grouping B has strong support only in MP analysis.
The controversy regarding the complex has been reported in previous studies (
In addition,

Patterns of shell colour within the genus Oxystele. A–C Oxystele variegata from Namibia, 5 km north of Swakopmund, diameter 22.2 mm (NMSA E6038) D–F Oxystele impervia from the Western Cape, Groen Rivier, diameter 22.3 mm (NMSA E7353) G–I Oxystele sp. from theEastern Cape, Tsitsikamma National Park, diameter 16.5 mm (HVDBM058-10, NMSA W7371); the colour pattern of these specimens suggests Oxystele variegata, but these specimens group within the unit of Oxystele impervia J–L Oxystele sp. from the Northern Cape, Noup, diameter 18.0 mm (HVDBM185-10, NMSA W7608); the colour pattern suggests Oxystele impervia, but they group with Oxystele variegata (see Figure 4 and Appendix 2 for the phylogenetic groupings of these specimens and node supports; these groupings contradict their morphological identification).
One of six polymorphic loci (glycyl-leucine peptidase or peptidase A;
Indeed morphological differentiation between both species can be difficult. Some of the shell colours and patterns are similar, and radula morphology could be altered as a result of differences in diet, age and other factors. For example,
In this study, most of the specimens that group within unexpected MOTUs were collected from different localities, suggesting possible shell colour variation due to variation in environmental conditions. For example, specimens of Oxystele variegata from Namibia and Robben Island clustered on the phylogeny, but those from north-western and southern Africa (Cape) did not. The Cape is renowned for its bad weather as indicated in its common name of “The Cape of Storms”, resulting in weathering of individuals (i.e. see “Ships in trouble in Cape waters”; http://www.e-gnu.com/shipwreck_update.html).
The split we found on the phylogeny and species delimitation analyses between Oxystele impervia and Oxystele variegata does not correspond with the nominal, morphologically-based identifications, indicating the need for the combination of morphological features and genetic data for further analysis. It is also possible that the COI gene alone is insufficient to discriminate species within the genus. We therefore suggest that future analysis should use a multi-gene approach. However,
We would like to thank the Government of Canada through Genome Canada and the Ontario Genomics Institute (2008-OGI-ICI-03) for the DNA sequencing. The research was supported by the ACDB and partially by the Toyota Enviro Outreach program 2010. The Tsitsikamma National Parks Board gave permission for sample collection and the Kwazulu-Natal Museum processed the voucher specimens. We thank Stephanus Voges and Bronwen Curry (Ministry of Fisheries and Marine Resources, Namibia), Estelle Esterhuizen (Robben Island Nature Conservation) and Gerhard Groenewald (Klipbokkop Nature Reserve) for assistance with sample collections. This work is based on research supported in part by the National Research Foundation of South Africa.
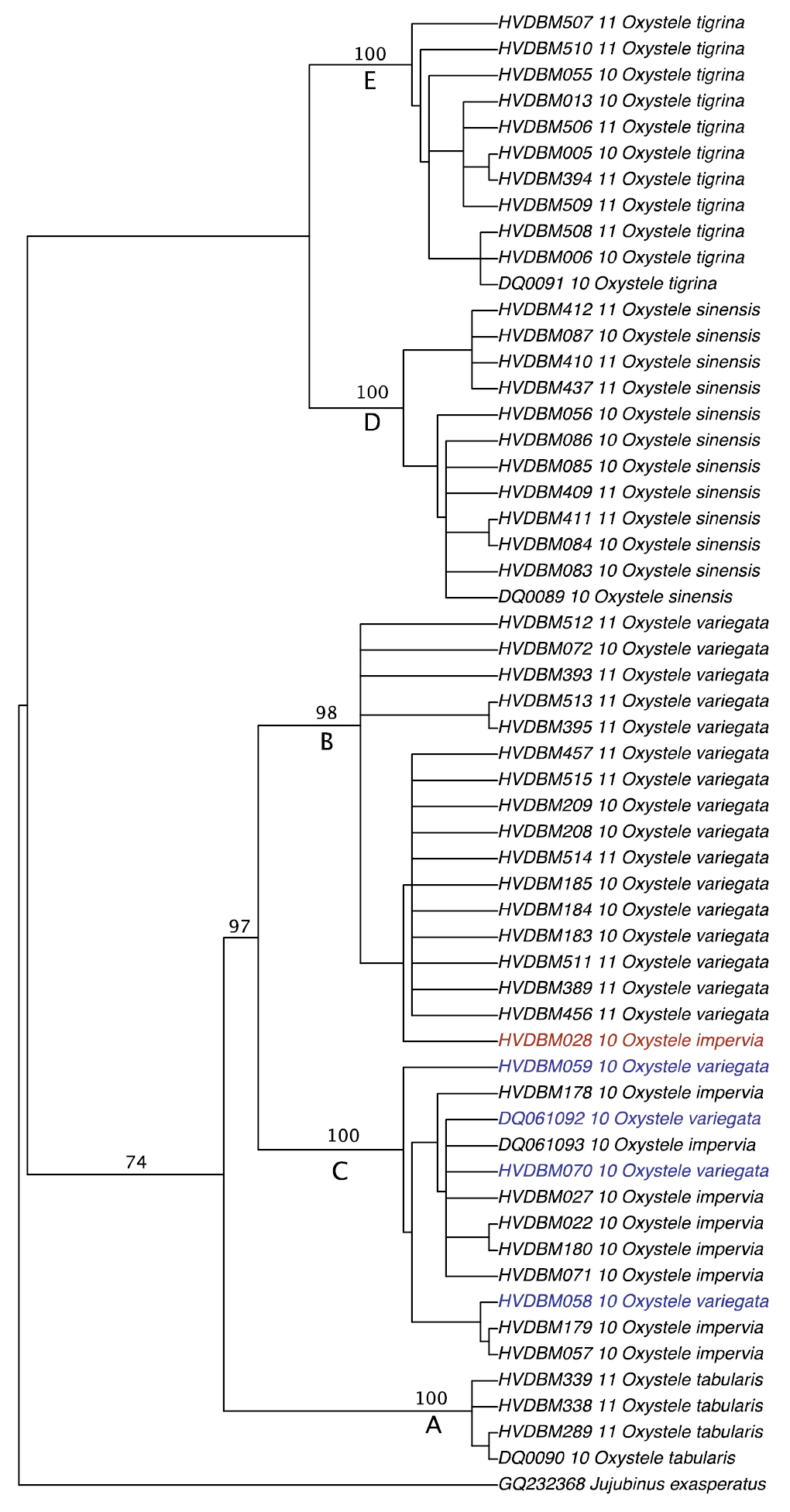
The only parsimonious tree obtained from the maximum parsimony (MP) analysis. Topology of species groupings is similar to that of the Bayesian tree (see Figure 3). Node supports are reported on the branches; the first value is bootstrap support from MP analysis; the second value in bracket indicates the posterior probability obtained from Bayesian analysis; only moderate to high node support values are indicated; Jujubinus exasperatus is used as outgroup; A-E indicates different possible species-units in the dataset: A (Oxystele tabularis), B (Oxystele variegata), C (Oxystele impervia), D (Oxystele sinensis), E (Oxystele tigrina), as in Figure 3.
